Here all all declarations of the global variables used throughout RNAlib. More...
 Include dependency graph for fold_vars.h:
Include dependency graph for fold_vars.h: This graph shows which files directly or indirectly include this file:
This graph shows which files directly or indirectly include this file:
Go to the source code of this file.
Functions | |
| void | set_model_details (model_detailsT *md) |
| Set default model details. More... | |
Variables | |
| int | fold_constrained |
| Global switch to activate/deactivate folding with structure constraints. | |
| int | noLonelyPairs |
| Global switch to avoid/allow helices of length 1. More... | |
| int | dangles |
| Switch the energy model for dangling end contributions (0, 1, 2, 3) More... | |
| int | noGU |
| Global switch to forbid/allow GU base pairs at all. | |
| int | no_closingGU |
| GU allowed only inside stacks if set to 1. | |
| int | tetra_loop |
| Include special stabilizing energies for some tri-, tetra- and hexa-loops;. More... | |
| int | energy_set |
| 0 = BP; 1=any mit GC; 2=any mit AU-parameter More... | |
| int | circ |
| backward compatibility variable.. this does not effect anything | |
| int | csv |
| generate comma seperated output | |
| int | oldAliEn |
| int | ribo |
| char * | RibosumFile |
| char * | nonstandards |
| contains allowed non standard base pairs More... | |
| double | temperature |
| Rescale energy parameters to a temperature in degC. More... | |
| int | james_rule |
| int | logML |
| int | cut_point |
| Marks the position (starting from 1) of the first nucleotide of the second molecule within the concatenated sequence. More... | |
| bondT * | base_pair |
| Contains a list of base pairs after a call to fold(). More... | |
| double * | pr |
| A pointer to the base pair probability matrix. More... | |
| int * | iindx |
| index array to move through pr. More... | |
| double | pf_scale |
| A scaling factor used by pf_fold() to avoid overflows. More... | |
| int | do_backtrack |
| do backtracking, i.e. compute secondary structures or base pair probabilities More... | |
| char | backtrack_type |
| A backtrack array marker for inverse_fold() More... | |
| int | gquad |
| Allow G-quadruplex formation. | |
| int | canonicalBPonly |
Detailed Description
Here all all declarations of the global variables used throughout RNAlib.
Function Documentation
| void set_model_details | ( | model_detailsT * | md | ) |
Set default model details.
Use this function if you wish to initialize a model_detailsT data structure with its default values, i.e. the global model settings
- See Also
- Parameters
-
md A pointer to the data structure that shall be initialized
Variable Documentation
| int noLonelyPairs |
Global switch to avoid/allow helices of length 1.
Disallow all pairs which can only occur as lonely pairs (i.e. as helix of length 1). This avoids lonely base pairs in the predicted structures in most cases.
| int dangles |
Switch the energy model for dangling end contributions (0, 1, 2, 3)
If set to 0 no stabilizing energies are assigned to bases adjacent to helices in free ends and multiloops (so called dangling ends). Normally (dangles = 1) dangling end energies are assigned only to unpaired bases and a base cannot participate simultaneously in two dangling ends. In the partition function algorithm pf_fold() these checks are neglected. If dangles is set to 2, all folding routines will follow this convention. This treatment of dangling ends gives more favorable energies to helices directly adjacent to one another, which can be beneficial since such helices often do engage in stabilizing interactions through co-axial stacking.
If dangles = 3 co-axial stacking is explicitly included for adjacent helices in mutli-loops. The option affects only mfe folding and energy evaluation (fold() and energy_of_structure()), as well as suboptimal folding (subopt()) via re-evaluation of energies. Co-axial stacking with one intervening mismatch is not considered so far.
Default is 2 in most algorithms, partition function algorithms can only handle 0 and 2
| int tetra_loop |
Include special stabilizing energies for some tri-, tetra- and hexa-loops;.
default is 1.
| int energy_set |
0 = BP; 1=any mit GC; 2=any mit AU-parameter
If set to 1 or 2: fold sequences from an artificial alphabet ABCD..., where A pairs B, C pairs D, etc. using either GC (1) or AU parameters (2); default is 0, you probably don't want to change it.
| int oldAliEn |
use old alifold energies (with gaps)
| int ribo |
use ribosum matrices
| char* RibosumFile |
warning this variable will vanish in the future ribosums will be compiled in instead
| char* nonstandards |
contains allowed non standard base pairs
Lists additional base pairs that will be allowed to form in addition to GC, CG, AU, UA, GU and UG. Nonstandard base pairs are given a stacking energy of 0.
| double temperature |
Rescale energy parameters to a temperature in degC.
Default is 37C. You have to call the update_..._params() functions after changing this parameter.
| int james_rule |
interior loops of size 2 get energy 0.8Kcal and no mismatches, default 1
| int logML |
use logarithmic multiloop energy function
| int cut_point |
Marks the position (starting from 1) of the first nucleotide of the second molecule within the concatenated sequence.
To evaluate the energy of a duplex structure (a structure formed by two strands), concatenate the to sequences and set it to the first base of the second strand in the concatenated sequence. The default value of -1 stands for single molecule folding. The cut_point variable is also used by PS_rna_plot() and PS_dot_plot() to mark the chain break in postscript plots.
| bondT* base_pair |
Contains a list of base pairs after a call to fold().
base_pair[0].i contains the total number of pairs.
- Deprecated:
- Do not use this variable anymore!
| double* pr |
A pointer to the base pair probability matrix.
- Deprecated:
- Do not use this variable anymore!
| int* iindx |
index array to move through pr.
The probability for base i and j to form a pair is in pr[iindx[i]-j].
- Deprecated:
- Do not use this variable anymore!
| double pf_scale |
A scaling factor used by pf_fold() to avoid overflows.
Should be set to approximately 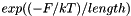 , where
, where 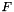 is an estimate for the ensemble free energy, for example the minimum free energy. You must call update_pf_params() after changing this parameter.
is an estimate for the ensemble free energy, for example the minimum free energy. You must call update_pf_params() after changing this parameter.
If pf_scale is -1 (the default) , an estimate will be provided automatically when computing partition functions, e.g. pf_fold() The automatic estimate is usually insufficient for sequences more than a few hundred bases long.
| int do_backtrack |
do backtracking, i.e. compute secondary structures or base pair probabilities
If 0, do not calculate pair probabilities in pf_fold(); this is about twice as fast. Default is 1.
| char backtrack_type |
A backtrack array marker for inverse_fold()
If set to 'C': force (1,N) to be paired, 'M' fold as if the sequence were inside a multi-loop. Otherwise ('F') the usual mfe structure is computed.
| int canonicalBPonly |
Do not use this variable, it will eventually be removed in one of the next versions
