Introduction
1.1 History
Microorganisms are essential for every part of human life—indeed all life on earth, they are found in almost every habitat present in nature. Even in hostile environments such as the poles, deserts, geysers, rocks, and the deep sea. They are vital to human health, ecology and other environment such as:- Soil health: A community if microorganisms is required to recycle nutrients. Farmers want to know what is there.
- Water pollution: Microorganism populations respond to the content of the water.
- Human health and nutrition: The microbial community of the gut, nose/throat, skin, vagina, are indicators of infected state, risk, and may be biomarkers of cancer.
- Paleobiology, Paleogenomics: DNA from frozen mammoth, iceman reveal their diet, phylogeny.
- Forensic science
One of the outcomes of examiating uncultured microorganisms is the appearance of "metagenomics". The term ‘metagenomics’ was first used by the Jo Handelsman group, and first appeared in publication in 1988, referring to the function-based analysis of mixed environmental DNA species. A new, and now most widely accepted meaning to the term has emerged as a result of the two works published in 2004 by Tyson and Venter, both describing the application of random whole genome shotgun sequencing to microbial populations. These two early studies defined the path for future metagenomic projects. Other terms have been used to describe the same method, including environmental DNA libraries, zoolibraries, soil DNA libraries, eDNA libraries, recombinant environmental libraries, whole genome treasures, community genome, whole genome shotgun sequencing, and probably others. "Metagenomics" is the most commonly used term.
At the same time, next generation sequencing enables metagenomics. Breakthroughs in developing alternative sequencing technologies occurred, promising a significantly higher throughput and considerably reduced cost of sequencing, thus providing the necessary platform for yet faster acquisition of metagenomic data. These new (known as next-generation) sequencing technologies, some of which are already widely utilized (the Roche 454, the Illumina Genome Analyzer and the Applied Biosystems SOLiD platforms) will be defining the future of metagenomics.
1.2 Definition
The basic definition of metagenomics is the analysis of genomic DNA from a whole community; this separates it from genomics, which is the analysis of genomic DNA from an individual organism or cell. Recently, metagenomics was defined as "the application of modern genomics techniques to the study of communities of microbial organisms directly in their natural environments, bypassing the need for isolation and lab cultivation of individual species".Metagenome data analysis aims at addressing at least one of the following questions:
- Diversity and abundance of community members ("who is there");
- Metabolic potential of the community and its members ("what they are doing");
- Ecological relations between members of the community ("why they are there").
1.3 Approaches to metagenomic analysis
Metagenomics can be divided into two key research areas, environmental single-gene surveys and random shotgun studies of all environmental genes. For the first one, single targets are amplified using the polymerase chain reaction (PCR), and then the products are sequenced, providing an analysis of the range of different orthologs (or paralogs) for that gene within a given community. Random shotgun metagenomics is a study in which total DNA has been isolated from a sample and then sequenced—resulting in a profile of all genes within the community. We annotate the genes and determine their relationship to the environment and then use this to identify proteins that can be synthesized by the metagenome.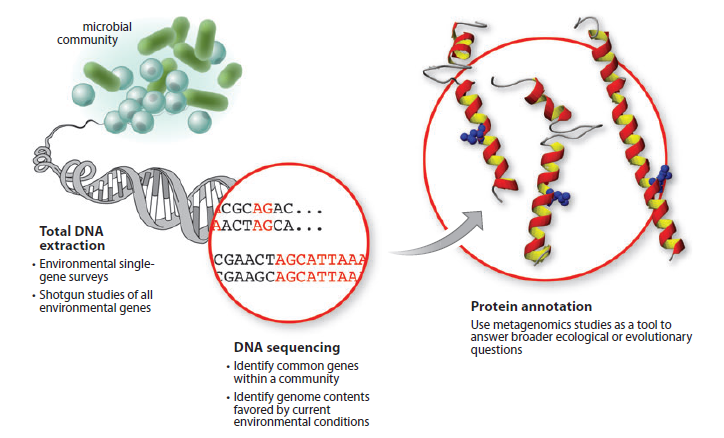
from: figure 1 of "Microbial metagenomics: beyond the genome."
The major steps in a metagenomic study include:- Sampling: Samples should represent the population from which they are taken. We need to obtain the environmental sample, filter the environmental sample and record the metadata;
- Sequencing: Shotgun sequencing and screens of clone libraries reveal genes present in environmental samples. This provides information both on which organisms are present and what metabolic processes are possible in the community. Shotgun metagenomics also is capable of sequencing nearly complete microbial genomes directly from the environment.
- Sequenc read preprocessing: Preprocessing of sequence reads prior to assembly, gene prediction and annotation is a critical and largely overlooked aspect of metagenomic analysis.
- Assembly: The reads are assembled into progressively longer contiguous sequences or contigs, and finally to the whole genome.
- Gene Prediction:Genes are the basic functional unit in the genome, which may constitute larger functional units such as operons, transcriptional units, and functional networks. The incomplete and fragmentary nature of metagenomic data presents challenges to identifying genes.
- Binning: We wish to know not only who populates the sample (gene prediction), but also what the different OTUs (Operational taxonomic unit, species distinction in microbiology) are doing. We must therefore associate sequence data with the OTU of its origin. This analysis is called binning.
- Funcational annotation: We would like to understand the functional potential of the microbial community from where we derived the metagenome. Metagenomics can be applied to solve practical challenges in medicine, engineering, agriculture, sustainability and ecology.
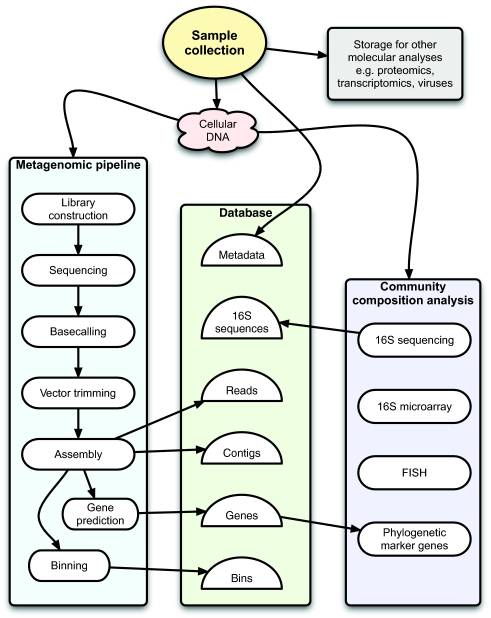
Typical workflow for Sanger-based metagenomic projects of bacterial and archaeal communities at the Joint Genome Institute (JGI). Oval boxes indicate processes, and half-circles indicate data. from: figure 1 of "A bioinformatician's guide to metagenomics."
Processing of metagenomic datasets, especially those derived from high-complexity microbiomes, is characterized by significantly higher error rate than processing of isolate genomes. The problems include:
- assembly of chimeric contigs (i. e. assembly of reads originating from different taxonomic groups)
- under-assembly (i. e. reads that should have been assembled remain as single-read contigs), higher rate of false-positive and false-negative results of gene prediction (mostly due to gene fragmentation)
- low sensitivity of binning (i. e. relatively small portion of scaffolds and contigs are assigned to bins, bins correspond to larger taxonomic groups than a species, etc.)
Reference:
1. Handelsman, Jo. "Metagenomics: application of genomics to uncultured microorganisms." Microbiology and Molecular Biology Reviews 68.4 (2004): 669-685..2. Riesenfeld, Christian S., Patrick D. Schloss, and Jo Handelsman. "Metagenomics: genomic analysis of microbial communities." Annu. Rev. Genet. 38 (2004): 525-552.
3. Chistoserdova, Ludmila. "Recent progress and new challenges in metagenomics for biotechnology." Biotechnology letters 32.10 (2010): 1351-1359.
4. Chen, Kevin, and Lior Pachter. "Bioinformatics for whole-genome shotgun sequencing of microbial communities." PLoS computational biology 1.2 (2005): e24.
5. Gilbert JA, Dupont CL., Microbial metagenomics: beyond the genome. Ann Rev Mar Sci. 2011;3:347-71.
6. Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. A bioinformatician's guide to metagenomics. Microbiol Mol Biol Rev. 2008 Dec;72(4):557-78, Table of Contents. doi: 10.1128/MMBR.00009-08.
7. Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Comput Biol. 2010 Feb 26;6(2):e1000667. doi: 10.1371/journal.pcbi.1000667.
8. Tyson, Gene W., et al. "Community structure and metabolism through reconstruction of microbial genomes from the environment." Nature 428.6978 (2004): 37-43.
9. Venter, J. Craig, et al. "Environmental genome shotgun sequencing of the Sargasso Sea." science 304.5667 (2004): 66-74.