Sampling and Sequencing
1.1 Sampling:
The first step in a metagenomic study is to obtain the environmental sample. The DNA extracted should be representative of all cells present in the sample and sufficient amounts of high-quality nucleic acids must be obtained for subsequent library production and sequencing.

Metadata are the 'data about the data': it includes detailed information about the three-dimensional (including depth, or height) geography and environmental features of the sample, physical data about the sample site, and the methodology of the sampling. There is a great need to have metadata in a standard, comprehensive and amenable way, as this may lead to biologically significant discoveries from statistically significant correlations analysis between the metagenomic data and the habitat-associated metadata.
1.2 Sequencing:
Over the past 10 years metagenomic shotgun sequencing has gradually shifted from classical Sanger sequencing technology to next-generation sequencing (NGS). There are two general sequencing strategies to obtain genome
sequence data from microbiome samples: directed sequencing
and shotgun sequencing of random clones.
Directed sequencing is either (i) function-driven, whereby clone libraries from a microbiome sample are sequenced after being screened for a desired
function; or (ii) driven by phylogenetic markers, whereby the
DNA flanking taxonomic anchors, such as 16S rDNA, is sequenced
in large-insert libraries.
Shotgun sequencing of microbiome
sample clone libraries follows a relatively unbiased
approach, which provides a broad survey of the gene content and
metabolic capabilities of a microbiome. Both sequencing methods have their advantages and disadvantages, as shown below:
| Method | Advantage | Disadvantage |
|---|---|---|
| Direct sequencing |
|
|
| Random shotgun sequencing |
|
|
A combination of shotgun and directed sequence approaches may emerge in the future and thus combine the advantages of the broad coverage provided by shotgun sequencing with the ability of sampling specific genome areas in low abundance organisms without over-sequencing more abundant members of the microbiome. Here our discussion pertains to metagenome data generated using shotgun sequencing. Shotgun metagenomics can be divided into the following categories:
- Fosmid, cosmid, and bacterial artificial chromosome (BAC)-derived metagenomic studies
- Sanger sequencing–derived shotgun metagenomic studies
- Next generation sequencing–derived shotgun metagenomic studies
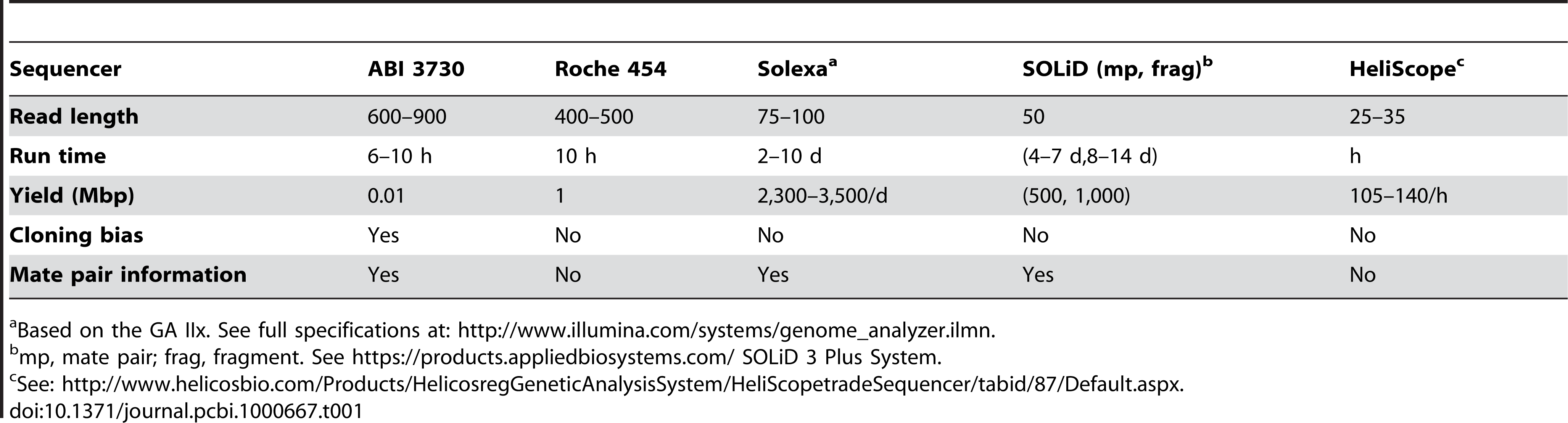
The following table shows a comparison between the yield, fragment length, and run times of the different sequencers.
1.3 Sequence Read Preprocessing:
Preprocessing comprises:
-
base calling of raw data coming off the sequencing machines:
Base calling is the procedure of identifying DNA bases from the readout of a sequencing machine. (tools: phred, Paracel's TraceTunner, ABI' KB, etc) -
vector screening to remove cloning vector sequence:
Vector screening is the process of removing cloning vector sequences from base-called sequence reads. (tools: cross_match, LUCK, vectro_clip, etc) - quality trimming to remove low-quality bases (as determined by base calling)
- contaminant screening to remove verifiable sequence contaminants
Reference:
1. Riesenfeld, Christian S., Patrick D. Schloss, and Jo Handelsman. "Metagenomics: genomic analysis of microbial communities." Annu. Rev. Genet. 38 (2004): 525-552.2. Thomas, Torsten, Jack Gilbert, and Folker Meyer. "Metagenomics-a guide from sampling to data analysis." Microb Inform Exp 2.3 (2012).
3. Prakash, Tulika, and Todd D. Taylor. "Functional assignment of metagenomic data: challenges and applications." Briefings in bioinformatics 13.6 (2012): 711-727.
4. Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Comput Biol. 2010 Feb 26;6(2):e1000667. doi: 10.1371/journal.pcbi.1000667.
5. Gilbert JA, Dupont CL., Microbial metagenomics: beyond the genome. Ann Rev Mar Sci. 2011;3:347-71.
6. Markowitz, Victor M., et al. "An experimental metagenome data management and analysis system." Bioinformatics 22.14 (2006): e359-e367.
7. https://dornsife.usc.edu/labs/laketyrrell/metagenomics/
8. Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. A bioinformatician's guide to metagenomics. Microbiol Mol Biol Rev. 2008 Dec;72(4):557-78, Table of Contents. doi: 10.1128/MMBR.00009-08.
9. Teeling, Hanno, and Frank Oliver Glöckner. "Current opportunities and challenges in microbial metagenome analysis—a bioinformatic perspective." Briefings in bioinformatics 13.6 (2012): 728-742.