Funcational annotation
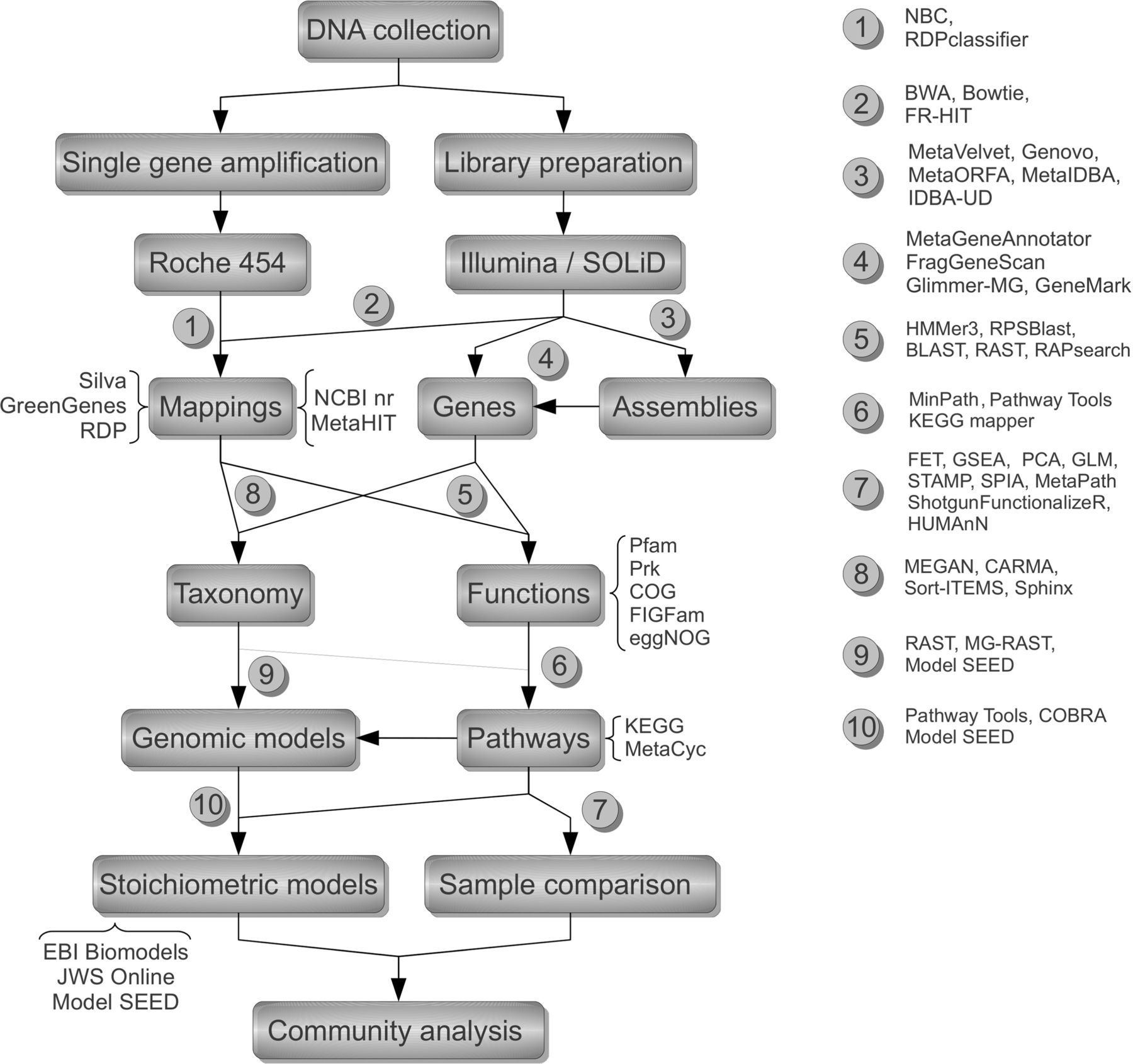
The analytic procedure ideally bifurcate at the starting point according to the investigation strategy: DNA can undergo a PCR-based amplification step to increase the amount of a specific marker gene (e.g. ribosomal RNA) and then subject to Roche 454 sequencing or can be fragmented and prepared into libraries for metagenomics Illumina/SOLiD sequencing. The simplest analytic choice is to map short reads into reference databases such as that maintained by the Ribosomal Database Project for the taxonomy survey via 16S sequencing (1) or into NCBI non-redundant (nr/nt) for environmental microbiome or, in case of gut microbiome surveys, the better-scoped MetaHIT (2). Another possibility is to assemble the short reads into longer contigs using new generation assemblers specific for unevenly distributed reads deriving from the multitude of different microbes represented in the community (3). Their application improves the efficiency of gene finding programs that, even though applicable directly on reads, have a higher level of information to ensure more confident gene identification (4). Once coding sequences have been obtained, their corresponding proteins can be searched in reference functional databases encoding information in the form of HMMs or PSSM from multiple sequence alignments (5) or directly in reference protein sets derived from primary databanks or from genome-derived collections. The first approach leads to a direct identification of associated functions that can be used to identify and score pathways (6) and in the end apply a battery of statistical techniques for sample characterization (7). The second approach can be used to obtain taxonomic and functional distributions (8) and allows to directly feed metabolic pathway identification (9) that in turn can be converted into stoichiometric models (10) for simulating the behaviour of single organisms or the relationships within a community, with the potential of predicting their response to changing environmental conditions.
Comparative analyses between metagenomes (comparative metagenomics) can provide additional insight into the function of complex microbial communities and their role in host health. Here is a list of commonly used pipelines for the functional annotation and comparison of metagenomic data sets.
| Year | Tools | Short Descriptions | URL |
|---|---|---|---|
| 2007 | CAMERA | The aim of this project is to serve the needs of the microbial ecology research community, and other scientists using metagenomics data, by creating a rich, distinctive data repository and a bioinformatics tools resource that will address many of the unique challenges of metagenomic analysis. | CAMERA |
| 2011 | CoMet | A web-server for fast comparative functional profiling of metagenomes. | CoMet |
| 2012 | HUMAnN | A pipeline for efficiently and accurately determining the presence/absence and abundance of microbial pathways in a community from metagenomic data. | HUMAnN |
| 2014 | IMG/M | Provides support for comparative analysis of microbial community aggregate genomes (metagenomes) in the context of a comprehensive set of reference genomes from all three domains of life, as well as plasmids, viruses and genome fragments. | IMG/M |
| 2014 | InterProScan | A tool that combines different protein signature recognition methods into one resource. | InterProScan |
| 2013 | MEGAN | Software for analyzing metagenomes. | MEGAN |
| 2011 | MetaPath | It can identify differentially abundant metabolic pathways in metagenomic datasets, relying on a combination of metagenomic sequence data and prior metabolic pathway knowledge (from KEGG). | MetaPath |
| 2010 | METAREP | An open source tool for high-performance comparative metagenomics. | METAREP |
| 2010 | MG-RAST | An automated analysis platform for metagenomes providing quantitative insights into microbial populations based on sequence data. | MG-RAST |
| 2009 | RAMMCAP | Analysis and comparison of very large metagenomes with fast clustering and functional annotation. | RAMMCAP |
| 2009 | ShotgunFunctionalizeR | An R-package for functional comparison of metagenomes. | ShotgunFunctionalizeR |
| 2010 | SmashCommunity | A stand-alone metagenomic annotation and analysis pipeline suitable for data from Sanger and 454 sequencing technologies. | SmashCommunity |
| 2010 | STAMP | A software package for analyzing metagenomic profiles that promotes ‘best practices’ in choosing appropriate statistical techniques and reporting results. | STAMP |
Reference:
1. De Filippo C, Ramazzotti M, Fontana P, Cavalieri D. "Bioinformatic approaches for functional annotation and pathway inference in metagenomics data." Brief Bioinform. 2012 Nov;13(6):696-710.