Assembly
Assembly is the process of combining sequence reads into contiguous stretches of DNA called contigs, based on sequence similarity between reads. The consensus sequence for a contig is either based on the highest-quality nucleotide in any given read at each position or based on majority rule.
Two strategies can be employed for metagenomics samples: reference-based assembly (co-assembly), and de novo assembly.
-
Reference based assembly works well, if the metagenomic dataset contains sequences where closely related reference genomes are available. However, differences in the true genome of the sample to the reference, such as a large insertion, deletion, or polymorphisms, can mean that the assembly is fragmented or that divergent regions are not covered.
-
De novo assembly typically requires larger computational resources. A whole class of assembly tools based on the de Bruijn graphs was specifically created to
handle very large amounts of data. Machine requirements for the de Bruijn assemblers are still significantly higher than for reference-based assembly (co-assembly).
|
The following table shows a list of commonly used tools for sequence assembly. Some of them are specific for metagenomic assembly. Metagenome assemblers differ from conventional genome assemblers in that they are designed for data containing more than one species, and so they generally have algorithms in place to separate species where possible, decreasing the amount of chimeric contigs constructed. They also tend not to rely on even coverage (the number of reads undering each consensus base is called depth or coverage) as a means of verifying assemblies unlike conventional genome assemblers, since coverage is not even in metagenomes because species have different abundances.
| Year | Tools | Short Descriptions | URL |
| 2002 | Arachne | Arachne was designed for long Sanger-chemistry reads. | Arachne |
| 2004 | Celera | Celera Assembler is a de novo whole-genome shotgun (WGS) DNA sequence assembler. It reconstructs long sequences of genomic DNA from fragmentary data produced by whole-genome shotgun sequencing. | Celera Assembler |
| 2007 | PHRAP | phrap is a program for assembling shotgun DNA sequence data. Among other features, it allows use of the entire read and not just the trimmed high quality part, it uses a combination of user-supplied and internally computed data quality information to improve assembly accuracy in the presence of repeats, it constructs the contig sequence as a mosaic of the highest quality read segments rather than a consensus, it provides extensive assembly information to assist in trouble-shooting assembly problems, and it handles large datasets. | PHRAP |
| 2008 | Velvet | Velvet is a de novo genomic assembler specially designed for short read sequencing technologies, such as Solexa or 454. | Velvet |
| 2010 | SOAPdenovo | SOAPdenovo is a novel short-read assembly method that can build a de novo draft assembly for the human-sized genomes. The program is specially designed to assemble Illumina GA short reads. | SOAPdenovo |
| 2011 | Genovo | Genovo uses a probabilistic model that calculates different coverage values to assemble metagenomes | Genovo |
| 2011 | Meta-IDBA | Meta-IDBA is an iterative De Bruijn Graph De Novo short read assembler specially designed for de novo metagenomic assembly. | Meta-IDBA |
| 2011 | Minimo | Minimo is designed to assemble small datasets and has been used for virome analyses | AMOS |
| 2012 | MetaVelvet | MetaVelvet is an extension of Velvet assembler to de novo metagenome assembly from short sequence reads | MetaVelvet |
| 2012 | IDBA-UD | IDBA-UD is a iterative De Bruijn Graph De Novo Assembler for Short Reads Sequencing data with Highly Uneven Sequencing Depth. It is an extension of IDBA algorithm. | IDBA-UD |
| 2012 | MAP | MAP is a de novo metagenomic assembly program for shotgun DNA reads. | MAP |
| 2012 | MOCAT | MOCAT is a metagenomics assembly and gene prediction toolkit. | MOCAT |
| 2012 | GeneStitch | GeneStitch is a novel way of using the de Bruijn graph assembly of metagenomes to improve the assembly of genes. | GeneStitch |
| 2012 | Ray Meta | Ray Meta is a massively distributed metagenome assembler that is coupled with Ray Communities, which profiles microbiomes based on uniquely-colored k-mers. |
Ray Meta |
| 2012 | VICUNA | VICUNA is a de novo assembly program targeting populations with high mutation rates. |
VICUNA |
| 2013 | MetAMOS | MetAMOS is a modular and open source metagenomic assembly and analysis pipeline. | MetAMOS |
| 2014 | GARM | GARM is (Genome Assembler, Reconcilation and Merging) a new software pipeline to merge and reconcile assemblies from different algorithms or sequencing technologies. | GARM |
| 2013 | PRICE | PRICE (Paired-Read Iterative Contig Extension) is a de novo genome assembler implemented in C++. | PRICE |
| 2013 | Xgenovo | Xgenovo generates quality assemblies with paired end reads. | Xgenovo |
| na | Newbler | Newbler is a software package for de novo DNA sequence assembly. It is designed specifically for assembling sequence data generated by the 454 GS-series of pyrosequencing platforms sold by 454 Life Sciences, a Roche Diagnostics company. | Newbler |
|
In all but the most species-poor metagenome, a full assembly is not possible:
-
The sampling is incomplete, and many if not all species’ genomes are partially sampled, if at all;
-
The species information itself is incomplete, and it is difficult to map individual reads to their species of origin.
-
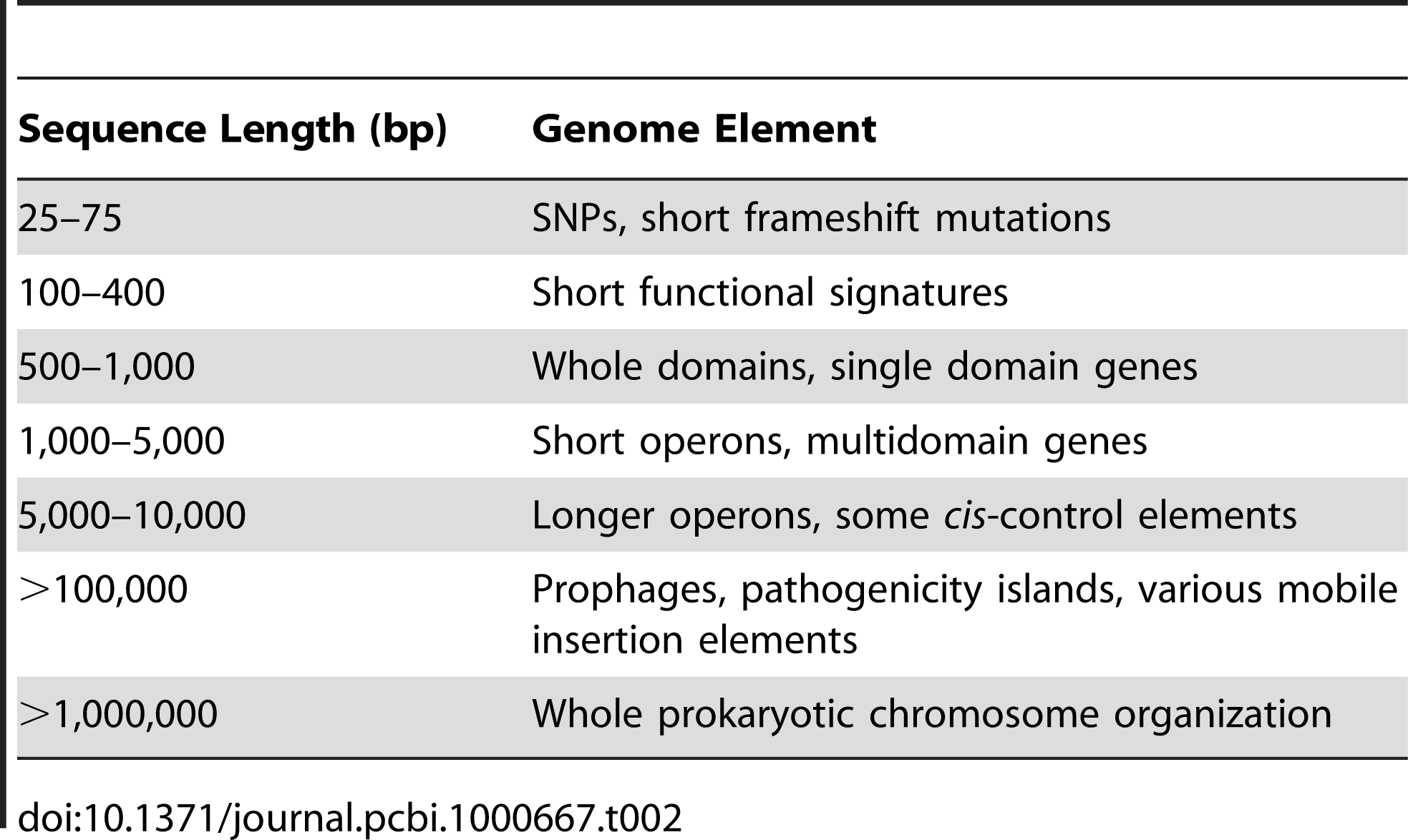
Analysis of genomic elements using metagenomic data is generally limited to the first three or four rows in the following Table.
The following table shows the information contained in different lengths of genomic DNA.
from: Table 2 of " A primer on metagenomics."
|
Some metagenomic assembling problems include:
-
Coverage (Coverage of a genome is defined as the mean
number of times a nucleotide is being sequenced.) is usually incomplete, since environmental sequence sampling rarely produces all the sequences required for assembly.
-
There is also the danger of assembling sequences from different OTUs, creating interspecies chimeras.
-
For short reads, they need to be produced in large quantities, and their short lengths means that there are many identical, or nearly identical, reads.
|
Other assembling problems are posed by the sequencing technologies as summarized in the following table.
| Sequencing technology feature | Assembly challenge |
| Short reads | Difficulty assembling repeats |
| Mate-pairs absent or difficult/expensive to obtain | Difficulty assembling repeats Lack of scaffolding information |
| New types of errors | Need to modify existing software and/or incorporate technology-specific features in assembly software |
| Large amounts of data (number of reads and size of auxiliary information) | Efficiency issues Require parallel implementations or specialized hardware when applied to large genomes |
Thus, new sequenicng and assembly technologies are expected to address all of these issures in the future.
|
Reference:
1. Vázquez-Castellanos, Jorge F., et al. "Comparison of different assembly and annotation tools on analysis of simulated viral metagenomic communities in the gut." BMC genomics 15.1 (2014): 37.
2. Prakash, Tulika, and Todd D. Taylor. "Functional assignment of metagenomic data: challenges and applications." Briefings in bioinformatics 13.6 (2012): 711-727.
3. Thomas, Torsten, Jack Gilbert, and Folker Meyer. "Metagenomics-a guide from sampling to data analysis." Microb Inform Exp 2.3 (2012).
4. Pop, Mihai. "Genome assembly reborn: recent computational challenges." Briefings in bioinformatics 10.4 (2009): 354-366.