MBMC V1.1 Manual
1. Prerequisites
MBMC can be run on systems (Linux, Windows) with Java installed:** Java 7 Runtime Environment.
You need to install Java 7 (64-bit is highly recommended). To check the version of the Java installed, please input the following command in the terminal:
java -versionThe latest version can be downloaded here.
Please make sure that java is available and can be accessed from the command prompt if you run MBMC on Windows, otherwise please add java path to the Environment variables. Be default, java will be installed in "c:\Program Files\Java\jre7\bin\java", so you can run java by using command as following:
For 32-bit java:
"c:\Program Files (x86)\Java\jre7\bin\java" -versionOr for 64-bit java:
"c:\Program Files\Java\jre7\bin\java" -version**Enough memory required
MBMC need enough memory to run. If the memory in your computer is limited, then it might run into the memory allocation error like:"Exception in thread "main" java.lang.OutOfMemoryError: Java heap space". We recommendate that MBMC should be run on a computer with at least 16GB memory.
** We use KAnalyze to do the k-mer counting.
This tool was included in the software package.
Reference: Audano, Peter, and Fredrik Vannberg. "KAnalyze: a fast versatile pipelined K-mer toolkit." Bioinformatics 30.14 (2014): 2070-2072.
** We need a pre-built database which contain 9-th order Markov Chains for each taxon.
This database was included in the software package.
2. Input Parameters
There are only three parameters for MBMC.-
Reads files (Required): paired-end or single-end reads in FASTA format
Paired-end reads should be in following format:
>Read1_end1
ACGT...
>Read1_end2
ACGT...
>Read2_end1
ACGT...
>Read2_end2
ACGT...
-
Cutoff c (Optional): the cutoff to determine the number of species, by default c=0.05.
This cutoff means that the binned group of reads which account for more than c of total reads will be regarded as a predicted species. -
Read type: 0 or 1 (Optional), by default Read type=0;
0: denotes paired-end reads;
1: denotes single-end reads;
3. Software Usage
We have both CLI (command line) and GUI versions for MBMC on Linux and Windows.(1) Command line versions (CLI) of MBMC
In terminal, use the following to run MBMC:
java -jar MBMC.jar -i reads_files -c 0.05 -r read_type(0 or 1)
- -i: reads files in FASTA format (Required)
- -c: cutoff to determine the number of species, by default c=0.05 (Optional)
- -r: 0: paired-end reads; 1: single-end reads, be default r=0 (Optional)
(2) GUI versions of MBMC
You can double-click the "MBMC_GUI.jar" executable to run it.
The following graph shows an example of GUI verions of MBMC:
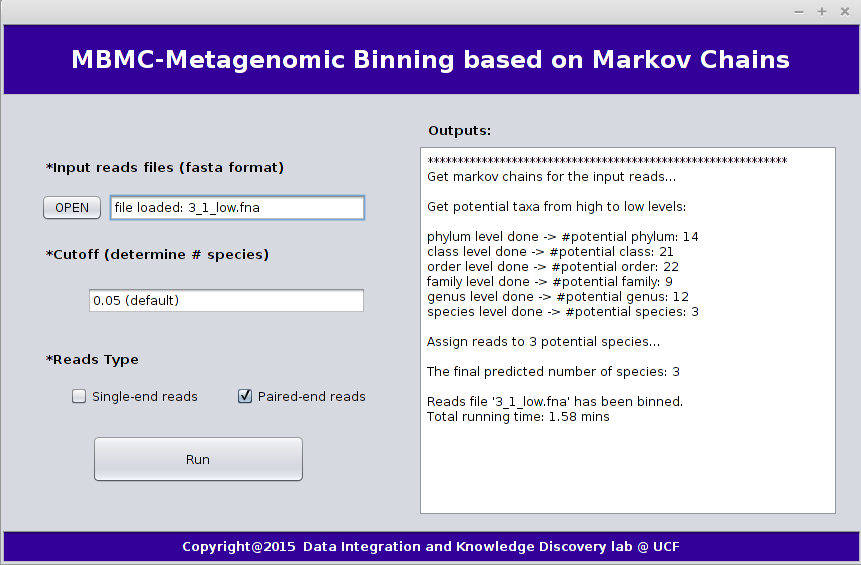
4. MBMC Results
We take dataset "3_1_low.fna" as an example, which is the smallest dataset in the MBMC paper. This dataset contains three species which were in the same order, the genome coverage for each species is 0.5 and abundance ratio is 1:1:1. We can run MBMC as the following command:java -jar MBMC.jar -i MBMC_examples/paired-end_reads/3_1_low.fna
(1) Output process:
This will give you a general look of the process when MBMC runs.
Example output process is as following:
************************************************************ Get markov chains for the input reads... Get potential taxa from high to low levels: phylum level done -> #potential phylum: 14 class level done -> #potential class: 21 order level done -> #potential order: 22 family level done -> #potential family: 9 genus level done -> #potential genus: 12 species level done -> #potential species: 3 Assign reads to 3 potential species... The final predicted number of species: 3 Reads file '3_1_low.fna' has been binned. Total running time: 1.58 mins(2) Output files:
Output files were binned reads in each of separate files in FASTA format, which can be found in the folder "ReadsFileName_binned".