|
Education Materials
Gene transcriptional regulation refers to any process by
which a cell regulates its genes expression. Properly regulated expression of
genes is crucial for ensuring that biological processes are accurately
carried out, for genes contributing to development, proliferation, programmed
cell death (apoptosis), aging, and differentiation. Gene expression begins
when mRNA molecules start to be synthesized, at the point on the gene where
they initiate. To understand the regulation of gene expression, it is
essential to discover the transcription initiation mechanisms under various
conditions, and how these varied mechanisms lead to different outcomes, or
phenotypes. High throughput sequencing of complete RNA sets synthesized in
cells has produced large datasets, but matching large-scale computational
studies, to understand phenotype-relevant transcription initiation mechanisms
are still at its early stage.
I. FlexSLiM: a Novel Approach for
Short Linear Motif Discovery in Protein Sequences
Short linear motifs (SLiMs) are 3
to 11 amino acid long peptide patterns that play important regulatory roles
in modulating protein activities. They Often occurs in protein disorder
regions, are high abundant in proteins. As an example, the FFAT SLiM, [DE].{0,4}E[FY][FYK]D[AC].[ESTD].
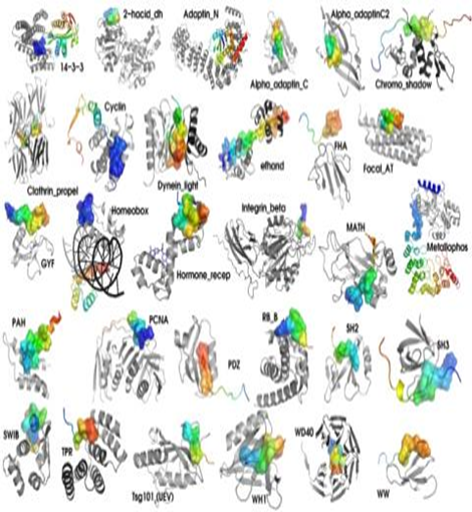
PLoS One, 2008, 3(7), e2524
Typical experiments involve raising an antibody to peptide
that expresses a SLiM and then using this antibody
to test the surface exposure or accessibility of the SLiM
(Nucleic Acids Res 26, 5486-5491, 1998), followed by the
mutation or deletion analysis. Although they are abundant in proteins, it is
often difficult to discover them by experiments, because of the low affinity
binding and transient interaction of short linear motifs with their partners.
Moreover, available computational methods cannot effectively predict short
linear motifs, due to their short and degenerate nature.
Effective methods are urgently needed to identify SLiMs. However, it is challenging to directly identify SLiMs, because a SLiM may
include a huge number of possible peptide patterns. For instance, the Fun_Delta SLiM [DE].{2,4}NN[IL] mentioned above
contains 2 × (20^2 + 20^3 + 20^4) × 1 × 1 × 2 = 673,600 different peptide
patterns with only fixed positions. We designate the peptide patterns
contained in a SLiM as SLiM
induced patterns, such as DRCNNI and D..NNI for the Fun_Delta SLiM. Because of the
large number of induced patterns contained in a SLiM,
many induced patterns are not statistically significant and it is thus also
difficult to identify all induced patterns directly. To resolve the above
issues, we propose to identify short elementary patterns first and then
combine elementary patterns into SLiMs.
By testing on simulated data and benchmark experimental
data, we demonstrated that FlexSLiM more
effectively identifies short linear motifs than existing methods. We provide
a general tool that will advance the understanding of short linear motifs,
which will facilitate the research on protein targeting signals, protein
post-translational modifications, and many others.

Lecture Slides Download Here.
2. Application
of Deep Learning Models to microRNA Transcription Start Site Identification
Micro-RNA (miRNA) refers to a class of
noncoding RNA that plays a role in post-transcriptional regulation. miRNA are
typically ~22 nucleotides in length and play a role in the down regulation of
the expression of more than 30% of mammalian gene products by binding to the
corresponding mRNA [1-3]. miRNAs regulate biological processes such as cell
differentiation, development, and apoptosis. Misexpression
of miRNAs has been associated with diseases such as diabetes, cancer, and
heart disease [4, 5]. Understanding the regulation and expression of miRNAs
is an essential component of understanding gene regulation and its role in
disease phenotypes. Transcription Start Sites (TSS)
are the locations within a promoter region where the transcription of gene
products begins. The TSSs of genes that produce
miRNAs are more difficult to study than their counterparts in genes that
produce proteins, due to the biogenesis process undergone by miRNAs [6, 7].
Long sequences of primary miRNAs (pri-miRNA) are
transcribed from the genes that ultimately produce mature miRNAs. The pri-miRNAs are then processed by nuclear RNase III Drosha and a cofactor protein to produce precursor miRNAs
(pre-miRNA). The pre-miRNAs are then cleaved by the RNase III Dicer to
produce RNA-induced Silencing Complexes (RISCs). The RISC, together with an
AGO protein, is involved to produce the mature miRNAs. The length of the pri-miRNAs relative to the mature miRNAs means that the TSSs for miRNAs can be surprisingly distant from the
mature miRNAs. In addition, the TSS biogenesis
process occurs so quickly that pri-miRNAs cannot be
captured in sufficient numbers by RNA-Seq
experiments. Because of these factors, it is difficult to identify the TSSs of miRNAs.
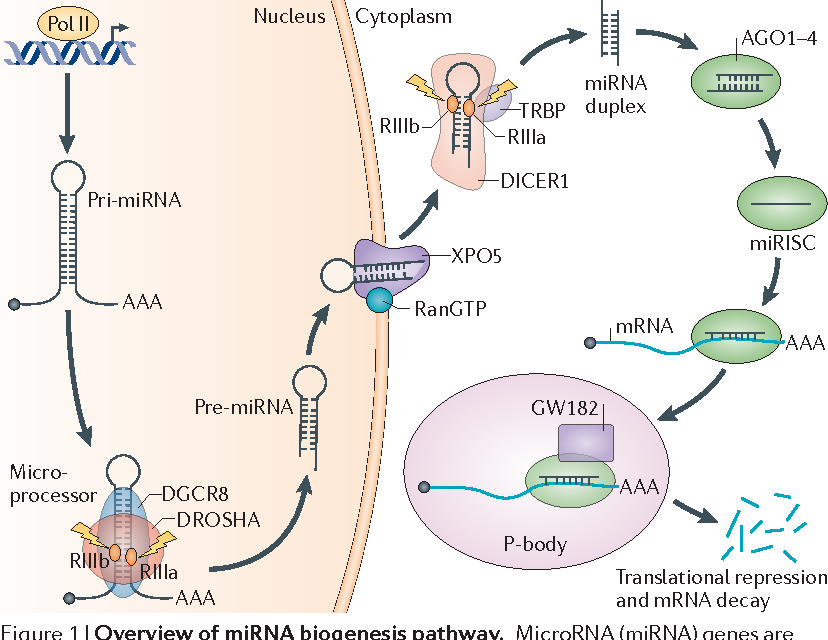
Lin S et al. Nature
Reviews Cancer 2015
We
have employed deep learning architectures incorporating Long Short-Term Memory
(LSTM) and Convolutional Neural Network (CNN) techniques to detect miRNA TSSs in regions of accessible chromatin. By testing on
benchmark experimental data, we demonstrated that deep learning models
outperform support vector machine and can accurately distinguish miRNA TSSs from both flanking regions and intergenic regions.
|